
In-Silico Comparative Analysis of Egyptian SARS CoV-2 with Other Populations: A Phylogeny and Mutation Analysis
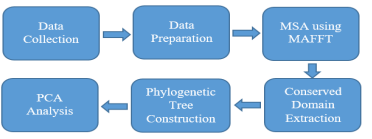
In the current SARS-CoV2 pandemic, identification and differentiation between SARS-COV2 strains are vital to attain efficient therapeutic targeting, drug discovery and vaccination. In this study, we investigate how the viral genetic code mutated locally and what variations is the Egyptian population most susceptible to in comparison with different strains isolated from Asia, Europe and other countries in Africa. Our aim is to evaluate the significance of these variations and whether they constitute a change on the protein level and identify if any of these variations occurred in the conserved domain of the virus. The available Covid-19 complete genome nucleotide sequences on NCBI were gathered and filtered, and representative sequences were selected from each of the mentioned continents to make the population of our sample 1535 sequences. Multiple sequence alignment was conducted for all the 1535 sequences obtained from NCBI. For higher accuracy, we used the MAFFT iterative refinement method. Conserved domain extraction was carried out for all 1535 sequence for mutation evaluation. When the mutations were evaluated, Spike_D614G, NSP12_P323L, NS3_Q57H and N_R203K were found to be the most common amino acid substitutions among the viral isolates from Egypt. All retrieved mutations were processed and analyzed with principal component analysis (PCA). In general, no clear clusters were clustered based on the mutation pattern of different continents, including Africa, Asia, and Europe. However, PCA shows that the African mutation pattern is a partial subset of the complete European mutation pattern. © 2020 IEEE.